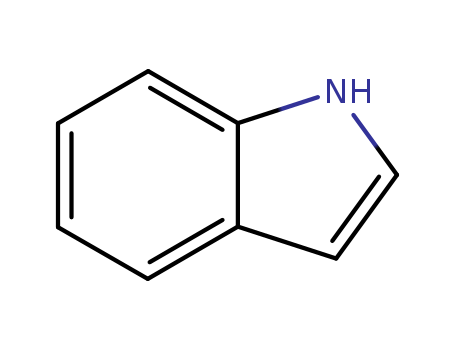
Indole literature
-
Suvorov et al.
, (1970)
-
One-pot tandem synthesis of 2,3-unsubstituted indoles, an improved Leimgruber-Batchoindole synthesis
Chen, Jinchun,Zhang, Zhikai,Liu, Sujing,Yang, Cuiyun,Xia, Chuanhai
, p. 4672 - 4675 (2014)
A concise, fast and efficient one-pot methodology has been developed for preparing 2,3-unsubstituted indoles from 2-nitrotoluenes and dimethylformamide dimethyl acetal. Compared with the classical Leimgruber-Batcho reaction, such a one-pot process simplified the operation procedures, generated less by-products and chemical residues, and resulted in higher overall yields in a shorter reaction time.
A BN Aromatic Ring Strategy for Tunable Hydroxy Content in Polystyrene
van de Wouw, Heidi L.,Lee, Jae Young,Awuyah, Elorm C.,Klausen, Rebekka S.
, p. 1673 - 1677 (2018)
BN 2-vinylnaphthalene, a BN aromatic vinyl monomer, is copolymerized with styrene under free radical conditions. Oxidation yields styrene–vinyl alcohol (SVA) statistical copolymers with tunable hydroxy group content. Comprehensive spectroscopic investigation provides proof of structure. Physical properties that vary systematically with hydroxy content include solubility and glass transition temperature. BN aromatic polymers represent a platform for the preparation of diverse functional polymeric architectures via the remarkable reaction chemistry of C?B bonds.
Properties of tryptophan indole-lyase from a piezophilic bacterium, Photobacterium profundum SS9
Phillips, Robert S.,Ghaffari, Rashin,Dinh, Peter,Lima, Santiago,Bartlett, Douglas
, p. 35 - 41 (2011)
Tryptophan indole-lyase (Trpase), PBPRA2532, from Photobacterium profundum SS9, a piezophilic marine bacterium, has been cloned, expressed in Escherichia coli, and purified. The P. profundum Trpase (PpTrpase) exhibits similar substrate specificity as the enzyme from E. coli (EcTrpase). PpTrpase has an optimum temperature for activity at about 30 °C, compared with 53 °C for EcTrpase, and loses activity rapidly (t1/2 ~ 30 min) when incubated at 50 °C, while EcTrpase is stable up to 65 °C. PpTrpase retains complete activity when incubated more than 3 h at 0 °C, while EcTrpase has only about 20% remaining activity. Under hydrostatic pressure, PpTrpase remains fully active up to 100 MPa (986 atm), while EcTrpase exhibits only about 10% activity at 100 MPa. PpTrpase forms external aldimine and quinonoid intermediates in stopped-flow experiments with l-Trp, S-Et-l-Cys, S-benzyl-l-Cys, oxindolyl-l-Ala, l-Ala and l-Met, similar to EcTrpase. However, with l-Trp a gem-diamine is observed that decays to a quinonoid complex. An aminoacrylate is observed with l-Trp in the presence of benzimidazole, as was seen previously with EcTrpase [28] but not with S-Et-l-Cys. The results show that PpTrpase is adapted for optimal activity in the low temperature, high pressure marine environment.
Origin of Stability and Inhibition of Cooperative Alkyne Hydrofunctionalization Catalysts
Chapple, Devon E.,Boyle, Paul D.,Blacquiere, Johanna M.
, p. 3789 - 3800 (2021)
New entries to the [Ru(Cp/Cp*)(PR2NR′2)(MeCN)]PF6 catalyst family were synthesized, including a Cp complex (R = Cy; R′ = Ph) and two Cp* complexes (R = Cy, Ph; R′ = Ph). These and other derivatives were used for the intramolecular hydroamination of 2-ethynylaniline to elucidate trends in catalytic lifetime and rate. The readily accessible [Ru(Cp)(PCy2NPh2)(MeCN)]PF6 derivative showed comparable lifetime to [Ru(Cp)(Pt?Bu2NPh2)(MeCN)]PF6, the previous optimal catalyst. Donor-free ‘active’ catalysts, [Ru(Cp/Cp*)(PCy2NPh2)]PF6, were prepared and their thermal stability was assessed. The relatively high stability of the Cp derivative was explained by the capacity of the PCy2NPh2 ligand to coordinate in a κ3-(P,P,Ar) mode, which protects the low-coordinate species. This coordination mode is inaccessible with the Cp* derivative. Additionally, [Ru(Cp*)(PCy2NPh2)]PF6 readily activated the C?Cl bond of the solvent dichloromethane. Variable time normalization analysis (VTNA) revealed that the indole product inhibited the catalyst [Ru(Cp)(PCy2NPh2)(MeCN)]PF6, which slowed catalytic rates.
back-to-Front Indole Synthesis Using Silver(I) Catalysis: Unexpected C-3 Pyrrole Activation Mode Supported by DFT
Clarke, Aimee K.,Lynam, Jason M.,Taylor, Richard J. K.,Unsworth, William P.
, p. 6844 - 6850 (2018)
An efficient silver(I)-catalyzed method is reported for the synthesis of substituted indoles, most notably 5-hydroxy-derivatives, via π-acidic alkyne activation. Most methods for the preparation of indoles involve annulation of a benzene precursor, but the method reported herein is unusual in that pyrrole precursors are used. Density Functional Theory (DFT) studies suggest that these reactions proceed via initial activation of the pyrrole C-3 position before undergoing subsequent rearrangement, contradicting the conventional wisdom that pyrroles are more nucleophilic through C-2.
Group VI metal-promoted endo-azacyclizations via alkyne-derived metal vinylidene carbenes
McDonald, Frank E.,Chatterjee, Arnab K.
, p. 7687 - 7690 (1997)
The molybdenum-promoted cycloisomerization of terminal alkynes tethered to nitrogen nucleophiles is described. Reaction of N-carbamoyl alkynylamines with (Et3N)Mo(CO)5 affords cyclic enecarbamates. Similarly, cyclization of 2-ethynylaniline gives the isomeric indole heterocycle, although N-3- butynylaniline affords the cyclic metal azacarbene product.
Ultrathin Amorphous/Crystalline Heterophase Rh and Rh Alloy Nanosheets as Tandem Catalysts for Direct Indole Synthesis
Ge, Jingjie,Yin, Peiqun,Chen, Ye,Cheng, Hongfei,Liu, Jiawei,Chen, Bo,Tan, Chaoliang,Yin, Peng-Fei,Zheng, Hong-Xing,Li, Qiang-Qiang,Chen, Shuangming,Xu, Wenjie,Wang, Xiaoqian,Wu, Geng,Sun, Rongbo,Shan, Xiang-Huan,Hong, Xun,Zhang, Hua
, (2021)
Heterogeneous noble-metal-based catalysis plays an essential role in the production of fine chemicals. Rh-based catalysts are one of the most active candidates for indole synthesis. However, it is still highly desired to develop heterogeneous Rh-based catalysts with high activity and selectivity. In this work, a general, facile wet-chemical method is reported to synthesize ultrathin amorphous/crystalline heterophase Rh and Rh-based bimetallic alloy nanosheets (NSs), including RhCu, RhZn, and RhRu. Impressively, the amorphous/crystalline heterophase Rh NSs exhibit enhanced catalytic activity toward the direct synthesis of indole compared to the crystalline counterpart. Importantly, the obtained amorphous/crystalline heterophase RhCu alloy NSs can further enhance the selectivity to indole of >99.9% and the conversion is 100%. This work demonstrates the importance of phase engineering and metal alloying in the rational design and synthesis of tandem heterogeneous catalysts toward fine chemical synthesis.
Ruthenium-catalyzed intramolecular hydroamination of aminoalkynes
Kondo, Teruyuki,Okada, Takumi,Suzuki, Toshiaki,Mitsudo, Take-Aki
, p. 149 - 154 (2001)
Low-valent ruthenium complexes with a π-acidic ligand, such as Ru(η6-cot)(dmfm)2 [cot=1,3,5-cyclooctatriene, dmfm=dimethyl fumarate] and Ru3(CO)12, showed high catalytic activity for the intramolecular hydroamination of aminoalkynes. The reaction is highly regioselective, in which a nitrogen atom is selectively attached to an internal carbon of alkynes to give five-, six-, and seven-membered nitrogen heterocycles as well as indoles in good to high yields.
A Reusable MOF-Supported Single-Site Zinc(II) Catalyst for Efficient Intramolecular Hydroamination of o-Alkynylanilines
Li, Beibei,Ju, Zhanfeng,Zhou, Mi,Su, Kongzhao,Yuan, Daqiang
, p. 7687 - 7691 (2019)
The exploitation of new and active earth-abundant metal catalysts is critical for sustainable chemical production. Herein, we demonstrate the design of highly efficient, robust, and reusable ZnII-bipyridine-based metal–organic framework (MOF) catalysts for the intramolecular hydroamination of o-alkynylanilines to indoles. Under similar conditions homogeneous catalytic systems mainly provide hydrolysate. Our results prove that MOFs support unique internal environments that can affect the direction of chemical reactions. The ZnII-catalyzed hydroamination reaction can be conducted without additional ligands, base, or acid, and is thus a very clean reaction system with regard to its environmental impact.
Protonated carbonic acid and reactive intermediates in the acidic decarboxylation of indolecarboxylic acids
Vandersteen, Adelle A.,Mundle, Scott O.C.,Kluger, Ronald
, p. 6505 - 6509 (2012)
Elucidation of the mechanism for decarboxylation of indolecarboxylic acids over a wide range of solution acidity reveals the importance of protonated carbonic acid (PCA) as a reaction intermediate. In concentrated acid, the initial addition of water to the carboxyl group of the indolecarboxylic acid leads to a hydrated species that is capable of releasing PCA upon rate-determining carbon-carbon bond cleavage. The overall process is catalytic in water and acid, implicating PCA as a potential carboxylating reagent in the microscopic reverse reaction.
A catalyst for the selective dehydrogenation of 4,5,6,7-tetrahydroindole into indole
Trofimov,Mikhaleva,Schmidt,Protsuk,Ivanov,Ryapolov
, p. 95 - 97 (2011)
-
DDQ as an electrocatalyst for amine dehydrogenation, a model system for virtual hydrogen storage
Luca, Oana R.,Wang, Ting,Konezny, Steven J.,Batista, Victor S.,Crabtree, Robert H.
, p. 998 - 999 (2011)
2,3-Dichloro-5,6-dicyanobenzoquinone (DDQ) is an electrochemical oxidation catalyst for a secondary amine, a model system for virtual hydrogen storage by removal of a hydrogen equivalent from an amine; a computational study provides mechanistic information. The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2011.
Conversion of Amines into Imines by Swern Oxidation
Keirs, David,Overton, Karl
, p. 1660 - 1661 (1987)
Indoline (1) and 2-methylindoline (2) have been converted into indole (3) and 2-methylindole (4), and dibenzylamine (7) and phenylbenzylamine (8) have been converted into the Schiff bases (9) and (10), by Swern oxidation; the methylthiomethyl amines (5), (6), (11), and (12) were the only other products formed.
Thermodynamics and kinetics of indole oligomerization: Preliminary results in aqueous sulfuric acid
Quartarone,Ronchin,Tortato,Vavasori
, p. 107 - 112 (2009)
Reaction rates and equilibrium constants of indole dimerization and trimerization in aqueous sulfuric acid at 298 K are reported. The equilibrium of oligomerization is attained in about 4-5 h, and formation of oligomers with more than three monomeric unit
From ascorbigens to indolocarbazoles
Preobrazhenskaya, Maria N.,Korolev, Alexander M.,Rozhkov, Ilya I.,Yudina, Larisa N.,Lazhko, Eduard I.,Aiello, Enrico,Almerico, Anna Maria,Mingoia, Francesco
, p. 265 - 274 (1999)
New methods of L-ascorbic acid derivatization with the use of polyfunctional indole-3-cabinols are described. Reaction of β-hydroxy-N-methyltryptamine and L-ascorbic acid gave lactame derivatives; (indol-3-yl)gylcolic and L-ascorbic acids produced 2-hydroxy-4-hydroxymethyl-3-(indol-3-yl)-)-cylopen-2-enone. Similarly 4-hydroxy-3-methoxyphenylglycolic and L-ascorbic acids yielded 2-hydroxy-3-(4-hydroxy-3-methoxyphenyl)-4-hydroxymethyl-cyclopen-2-enone. Properties of N-methoxyascorbigen (neoascorbigen) were investigated. Alkylation of L-ascorbic acid with polysubstituted pyrrolecarbinols led to pyrrole analogues of ascorbigen. Acidic transformation of 3-formylindole and 1-methyl-3-formylindole led to indolocarbazoles and triindolylmethane derivatives.
Nucleophilic dimerization of indoline under oxidative conditions
Kovalev, Igor S.,Kopchuk, Dmitry S.,Zyryanov, Grigory V.,Rusinov, Vladimir L.,Chupakhin, Oleg N.
, p. 40 - 41 (2014)
Oxidation of indoline with 30% hydrogen peroxide in methanol in the presence of sodium tungstate affords the dimeric 3-oxo-1'H,3H- 2,3'-biindole-1-oxide.
-
Panunzio et al.
, p. 415 (1972)
-
Simultaneous chemosensing of tryptophan and the bacterial signal molecule indole by boron doped diamond electrode
Buzid, Alyah,Reen, F. Jerry,O'Gara, Fergal,McGlacken, Gerard P.,Glennon, Jeremy D.,Luong, John H.T.
, p. 845 - 852 (2018)
A simple and robust chemosensing approach using a boron-doped diamond (BDD) electrode has been developed and applied to analyze tryptophan (TRP) and indole during the growth of Escherichia coli in a complex growth medium. The bacterial enzyme tryptophanase catalyzes TRP to indole, an emerging signaling molecule. The process can now be monitored using electrochemistry, in a method far beyond the traditional identification protocols. Electroanalysis in a non-aqueous medium comprising acetonitrile (ACN) and tetrabutylammonium hexafluorophosphate (TBAH) is capable of separating the oxidation peak of TRP from that of indole. Mechanisms are postulated for the electrochemical oxidation of indole and TRP in ACN chosen because of its wider potential range, proton acceptor property, and solubilization of analytes. The electrochemical oxidation of TRP involves the elimination of two electrons. With a detection limit of 0.5 μM for both indole and TRP, this chemosensing approach is sufficient to monitor the level of these two biomolecules during the bacterial growth period.
CYCLIC ALLYLAMINE/ENAMINE SYSTEMS-6 SOME REACTIONS OF 4-(INDOL-2-YLCARBONYL)-, 4-(INDOL-3-YLCARBONYL)- AND 4-(INDOL-3-YLMETHYL)-1,2,5,6-TETRAHYDRO-1-METHYLPYRIDINES
Martinez, Silvio J.,Dalton, Lesley,Joule, John A.
, p. 3339 - 3344 (1984)
Indol-3-yl 1-methyl-1,2,5,6-tetrahydropyridin-4-yl ketone (1d) can be isomerised to indol-3-yl 1-methyl-1,2,3,4-tetrahydropyridin-4-yl- ketone but the protonated form of this enamine could not be cyclised to the indole α-position.Both indol-2-yl 1-methyl-1,2,5,6-tetrahydropyridin-4-yl ketone (1c) and its isomer (1d) were cyclised to 5-membered ketones by mineral acid catalysed Michael-type addition of indole β- and α-positions respectively onto the unsaturated ketone systems.Ketone (1d) was transformed to 1-acetylindol-3-yl 3-acetyl-1,4,5,6-tetrahydropyridin-4-yl ketone by hot acetic anhydride.Strong base treatment of indol-3-yl(1,2,5,6-tetrahydropyridin-4-yl) methane caused isomeration of the double bond into conjugation with the indole rather than into the endocyclic enamine position.
Evolution of the indole alkaloid biosynthesis in the genus Hordeum: Distribution of gramine and DIBOA and isolation of the benzoxazinoid biosynthesis genes from Hordeum lechleri
Gruen, Sebastian,Frey, Monika,Gierl, Alfons
, p. 1264 - 1272 (2005)
Two indole alkaloids with defense related functions are synthesized in the genus Hordeum of the Triticeae. Gramine (3(dimethyl-amino-methyl)-indole) is found in H. spontaneum and in some varieties of H. vulgare, the benzoxazinoid 2,4-dihydroxy-2H-1,4-benzoxazin-3(4H)-one (DIBOA) is detected in H. roshevitzii, H. brachyantherum, H. flexuosum, H. lechleri. Biosynthesis of DIBOA and of gramine was found to be mutually exclusive in wild Hordeum species, indicating that there was selection against simultaneous expression of both pathways during evolution. The full set of genes required for DIBOA biosynthesis in H.lechleri was isolated and the respective enzyme functions were analyzed by heterologous expression. The cytochrome P450 genes Bx2-Bx5 demonstrate a monophyletic origin for H. lechleri, Triticum aestivum and Zea mays. HlBx2-HlBx5 share highest homology to the orthologous genes of T. aestivum. In contrast, the branch point enzyme of the DIBOA pathway, the indole-3-glycerol phosphate lyase BX1, might have evolved independently in H. lechleri. In all Hordeum species that synthesize DIBOA, DNA sequences homologous to Bx genes are found. In contrast, these sequences are not detectable in the genomes of H. vulgare and H. spontaneum that do not synthesize benzoxazinoids.
Acceptorless dehydrogenation of N-heterocycles by supported Pt catalysts
Moromi, Sondomoyee K.,Siddiki,Kon, Kenichi,Toyao, Takashi,Shimizu, Ken-ichi
, p. 507 - 511 (2017)
Pt metal nanoparticles loaded on various supports and carbon-supported various metal catalysts are tested for dehydrogenation of 6-methyl-1,2,3,4- tetrahydroquinoline to 6-methyl-quinoline under oxidant-free conditions. In the 20 types of the catalysts screened, carbon-supported Pt catalyst (Pt/C) shows the highest activity. Pt/C is reusable after the reaction and is effective for dehydrogenation of various N- heterocycles (tetrahydroquinolines and indoline). Pt/C is also effective for hydrogenation of quinoline under 3 bar H2. The results demonstrate that this catalytic method may be useful for an organic hidride–based hydrogen storage system.
Reaction pathway in the vapour-phase synthesis of indole and alkylindoles
Campanati,Franceschini,Piccolo,Vaccari
, p. 1 - 9 (2005)
The vapour-phase synthesis of indole and its derivatives from aniline or alkylanilines and ethylene glycol or other diols was investigated with the use of a novel ZrO2/SiO2 (5:95 w/w) catalyst to check the applicability of this synthesis to a wide number of alkylindoles. During feeding with alkylaniline, the above catalyst showed catalytic results better than those reported in the literature, and a very good regenerability. In particular, with ethylene glycol, the best yields in the corresponding indoles were obtained when a C2-C3 alkyl chain was located in the ortho position to the amino group. The differences in reactivity between aniline and alkylaniline were significantly reduced when the length of the diol chain was increased and eliminated with 2,3-butanediol. On the basis of the above data and those collected sharing the synthesis in single steps, a possible overall reaction pathway was proposed to design a better tailor-made catalyst. It was also indicated that the formation of heavy compounds, which are able to deactivate the catalyst, were not derived from the reagents or the following reactions on the indole formed, but might be mainly attributed to the polycondensation of an aldehyde intermediate.
Electron Transfer From Indoles, Phenol, and Sulfite (SO32-) to Chlorine Dioxide (ClO2.)
Merenyi, Gabor,Lind, Johan,Shen, Xinhua
, p. 134 - 137 (1988)
With the ClO2/ClO2- couple as reference the one-electron-reduction potentials have been determined for four methylated indolyl radical cations.Their Eo values are 1.23 V (N-Me), 1.10 V (2-Me), 1.07 V (3-Me), and 0.93 V (2,3-diMe).Eo values were also measured for the following: tryptophylH.+/trypH 1.24 V, SO3.-/SO32- 0.76 V, and phenoxy./phenolate 0.80 V.The redox potentials were obtained from purely kinetic data (for tryptophan and 2-, 3-, and N-methylindole) or from combined kinetic and thermodynamic measurements.
Radicals and radical ions derived from indole, indole-3-carbinol and diindolylmethane
Bloch-Mechkour, Anna,Bally, Thomas,Sikora, Adam,Michalski, Radoslaw,Marcinek, Andrzej,Gebicki, Jerzy
, p. 6787 - 6794 (2010)
The primary products, i.e., the radical cations and radicals obtained on oxidation of the glucobrassicin metabolites (and dietary supplements), indole-3-carbinol (I3C) and diindolylmethane (DIM), and those from parent indole (I) are characterized in an ionic liquid and in Ar matrices. The radical cations of I and I3C are stable toward (photo)deprotonation under these conditions, but the resulting radicals can be generated by UV-photolysis of the neutral precursors. Two types of radicals, obtained by loss of hydrogen from N- and C-atoms, respectively, are found for I3C and DIM.
-
Bergman et al.
, p. 3347,3350,3351 (1970)
-
Transition metal-free regioselective C-3 amidation of indoles with N-fluorobenzenesulfonimide
Liu, Hai-Hong,Wang, Yi,Deng, Guojun,Yang, Luo
, p. 3369 - 3374 (2013)
A direct transition metal-free regioselective C-3 amidation of indoles has been developed with the commercially available N-fluorobenzenesulfonimide (NFSI) as the amino source under external oxidant-free conditions. This amidation requires only a catalytic amount of base and exhibits excellent functional group tolerance and regioselectivity. The C-3 regioselectivity was proposed to realize by a free radical mechanism. Copyright
Refractometric studies of molecular complexes of DDT with some compounds of biological interest
Sahai,Chauhan,Singh
, p. 935 - 943 (1981)
-
Striking effects of a titania support on the low-temperature activities of Ir catalysts for the dehydrogenative synthesis of benzimidazole and indole
Fukutake, Tatsuhiro,Wada, Kenji,Liu, Gang Chuan,Hosokawa, Saburo,Feng, Qi
, p. 235 - 240 (2018)
The crystalline structure of titania supports for iridium catalysts markedly affected their low-temperature activity for the dehydrogenative synthesis of N-containing heteroaromatics, namely benzimidazole and indole. While solid iridium catalysts supported on anatase showed moderate to poor activity for the synthesis of 2-phenylbenzimidazole (3) from o-phenylenediamine (1) and benzyl alcohol (2) at 100 °C, the reaction in the presence of rutile-supported catalysts proceeded smoothly to give 3 in high yields of up to 88%. Similar results were observed for the dehydrogenative conversion of 2-(2-aminophenyl)-ethanol (4) to indole (5). The reaction at 100 °C for 18 h in the presence of 1.0 mol% iridium on rutile gave 5 in a yield of 73%, while the use of anatase-supported catalysts resulted in significantly lower yields. TEM analysis showed the formation of small (ca. 2 nm in diameter), homogeneously-dispersed iridium nanoparticles on rutiles, while the inhomogeneous loading of iridium species was observed on anatase supports. CO pulse experiments revealed that there is a strong correlation between CO uptake by iridium nanoparticles and the activities at 100 °C. These results suggest that the predominant formation of small, well-reduced iridium nanoparticles is one major reason for the excellent activities of rutile-supported catalysts at low temperatures.
The Catalytic Life of CdBr2-KBr and Its Affect on the Rate of Indole Formation from Aniline and Ethylene Glycol
Seto, Takatoshi,Kujira, Katsufumi,Iwane, Hiroshi,Imanari, Makoto
, p. 3665 - 3670 (1995)
In the liquid-phase synthesis of indole from aniline and ethylene glycol (EG), a CdBr2-KBr catalyst gave a high yield of indole.In recycling experiments using the catalyst in an autoclave, the yield of indole remained stable and the inorganic comonents of the catalysts did not suffer degeneration.The time course of the reaction under pressure and high temperature was evaluated using a newly devised autoclave.The rate of formation of indole was determined to be proportional to the concentrations of both EG and the catalyst.Based on these findings for the reaction over CdBr2-3KBr, it is clear that this catalyst provides a simple and low-cost catalytic process for the formation of indole.
-
Ermolenko et al.
, (1978)
-
-
Tyson
, p. 2801 (1950)
-
OXIDATIVE DECARBOXYLATION OF CYCLIC AMINO ACIDS AND DEHYDROGENATION OF CYCLIC SECONDARY AMINES WITH IODOSOBENZENE
Ochiai, Masahito,Inenaga, Minako,Nagao, Yoshimitsu,Moriarty, Robert M.,Vaid, Radhe K.,Duncan, Michael P.
, p. 6917 - 6920 (1988)
Cyclic amino acids L-proline, pipecolinic acid and L-2-pyrrolidinone-5-carboxylic acid undergo oxidative decarboxylation with iodosobenzene in various solvents (including water) to yield the lactam and imide in the latter case.The reaction proceeds via initial imine formation.
2,2,6,6-Tetramethylpiperidin-1-yloxycarbonyl: A Protecting Group for Primary, Secondary, and Heterocyclic Amines
Lizza, Joseph R.,Bremerich, Maximilian,McCabe, Stephanie R.,Wipf, Peter
, p. 6760 - 6764 (2018)
The 2,2,6,6-tetramethylpiperidin-1-yloxycarbonyl (Tempoc) protecting group is readily introduced by the reaction of amines with a new acyl transfer reagent, 4-nitrophenyl (2,2,6,6-tetramethylpiperidin-1-yl) carbonate (NPTC). Tempoc has a reactivity profile that complements the commonly used t-butyloxycarbonyl (Boc) and benzyloxycarbonyl (Cbz) protecting groups. Deprotection can be achieved under mild reductive conditions with in situ generated Cu(I) species or by thermolytic cleavage at 135 °C. Mechanistic studies on the deprotection of Tempoc-indole suggest a combination of ionic and radical fragmentation pathways under thermal conditions.
SUBSTITUTION NUCLEOPHILE RADICALAIRE (SRN1) INDUITE PAR VOIE ELECTROCHIMIQUE
Boujlel, K.,Simonet, J.,Roussi, G.,Beugelmans, R.
, p. 173 - 176 (1982)
The electrochemical method is used for initiate a radical nucleophilic substitution leading to the synthesis of indoles.
NH4Cl-promoted synthesis of symmetrical and unsymmetrical triindolylmethanes under solvent-free conditions
Naskar, Subhendu,Hazra, Abhijit,Priyankar Paira,Sahu, Rishnendu B.,Banerjee, Sukdeb,Mondal, Nirup B.
, p. 568 - 571 (2008)
The synthesis of various triindolylmethanes from indole-3-carboxaldehyde, using indole derivatives as reactants and NH4Cl as catalyst under solvent-free conditions, is described. This methodology provides access to both symmetrical and unsymmetrical triindolylmethanes in excellent yields. With N-methylindole particularly, indole-3-carboxaldehyde appears to act as a formyl donor, leading to the exclusive formation of a symmetrically trisubstituted product. The novelty of the methodology lies in its operational simplicity, environment friendly reaction conditions, and inexpensive and easy availability of the catalyst. A plausible mechanism of formation of the products is suggested.
Pyrylenes: A New Class of Tunable, Redox-Switchable, Photoexcitable Pyrylium-Carbene Hybrids with Three Stable Redox-States
Antoni, Patrick W.,Hansmann, Max M.
, p. 14823 - 14835 (2018)
A new synthetic and modular access to a large family of redox-switchable molecules based upon the combination of pyrylium salts and carbenes is presented. The redox-properties of this new molecule class correlate very well with the π-accepting properties of the corresponding carbenes. While the pyrylium moiety acts as a chromophore, the carbene moiety can tune the redox-properties and stabilize the corresponding radicals. This leads to the isolation of the first monomeric pyranyl-radical in the solid-state. The three stable oxidation states could be cleanly accessed by chemical oxidation, characterized by NMR, EPR, UV-vis, and X-ray diffraction and supported by (TD)-DFT-calculations. The new hybrid class can be utilized as an electrochemically triggered switch and as a powerful photoexcited reductant. Importantly, the pyrylenes can be used as novel photocatalysts for the reductive activation of aryl halides and sulfonamides by consecutive visible light induced electron transfer processes.
Proton transfer and carbon-carbon bond cleavage in the elimination of indole catalyzed by Escherichia coli tryptophan indole-lyase
Phillips, Robert S.,Sundararaju, Bakthavatsalam,Faleev, Nicolai G.
, p. 1008 - 1014 (2000)
Tryptophan indole-lyase from Escherichia coli catalyzes the reversible cleavage of L-tryptophan to indole and ammonium pyruvate. This reaction is mechanistically interesting since it involves the elimination of an aromatic carbon leaving group. We have been studying the mechanism of tryptophan indole-lyase using rapid-scanning stopped-flow spectrophotometry. Recently, we demonstrated that the rate constant for α-aminoacrylate intermediate formation from α-2H-L-tryptophan exhibits an isotope effect of 3.0 (Sloan, M. J.; Phillips, R. S. Biochemistry 1996, 35, 16165-16173). We have confirmed this previous result (Dk = 2.99 ± 0.30) and we have now found that β,β-di-2H-L-tryptophan also exhibits a secondary isotope effect (Dk = 1.17 ± 0.03) on the elimination reaction. Furthermore, α,β,β-tri-2H-L-tryptophan exhibits a multiple isotope effect (Dk = 4.42 ± 0.67) on the elimination of indole. In addition, there is a significant solvent isotope effect (Dk = 1.79 ± 0.11) on indole elimination in D2O. This solvent isotope effect combines with the effect of α-deuterium, since elimination of α-2H-L-tryptophan in D2O exhibits Dk = 4.30 ± 0.16. In addition, the rate constant for indole elimination shows a linear Eyring plot between 5 and 35 °C. In the direction of tryptophan synthesis, the reaction of the α-aminoacrylate intermediate with indole to form a quinonoid intermediate also exhibits a kinetic isotope effect for 3-2H-indole, with Dk = 1.88 ± 0.19. In contrast to our expectations, the results suggest that the proton transfer and carbon-carbon bond cleavage in the elimination reaction are very nearly simultaneous and that the indolenine structure is a transient intermediate which occupies a very shallow well on the reaction coordinate, or a transition state, in the reaction of Trpase.
A NEW DEHYDROGENATION REACTION OF INDOLINES TO INDOLES VIA AZASULFONIUM SALTS
Kikugawa, Yasuo,Kawase, Masami
, p. 445 - 446 (1981)
Indolines (1) have been converted to the corresponding azasulfonium salts (2) and the subsequent intramolecular base catalyzed abstraction of the hydrogen at C-2 gave indoles (4) in good yields.
Indole synthesis by palladium-catalyzed tandem allylic isomerization-furan Diels-Alder reaction
Xu, Jie,Wipf, Peter
, p. 7093 - 7096 (2017)
A Pd(0)-catalyzed elimination of an allylic acetate generates a π-allyl complex that is postulated to initiate a novel intramolecular Diels-Alder cycloaddition to a tethered furan (IMDAF). Under the reaction conditions, this convergent, microwave-accelerated cascade process provides substituted indoles in moderate to good yields after Pd-hydride elimination, aromatization by dehydration, and in situ N-Boc cleavage.
Kinetics and mechanism of the basic hydrolysis of indomethacin and related compounds: A reevaluation
Cipiciani,Ebert,Linda,Rubessa,Savelli
, p. 1075 - 1076 (1983)
The kinetics of the hydrolysis of indomethacin and related compounds were studied in an alkaline medium at 25°. The pseudo-first-order rate constants were evaluated from log absorbance versus time plots in the ultraviolet. These compounds showed a second-order rate constant at low concentrations of hydroxide ion and a first-order rate constant at higher concentrations of hydroxide ion.
-
Nielsen et al.
, p. 127 (1978)
-
Improved indole syntheses from anilines and vicinal diols by cooperative catalysis of ruthenium complex and acid
Zhang, Min,Xie, Feng,Wang, Xiaoting,Yan, Fengxia,Wang, Ting,Chen, Mengmeng,Ding, Yuqiang
, p. 6022 - 6029 (2013)
By developing a new and efficient dinuclear catalyst [Ru(CO) 2(Xantphos)]2 [Xantphos = 4,5-bis(diphenylphosphino)-9,9- dimethyl-9H-xanthene], an improved synthesis of indole from vicinal diols and anilines by cooperative catalysis of ruthenium complex and p-TSA (para-toluenesufonic acid) has been demonstrated. The presented synthetic protocol allows assembling a wide range of products in an efficient manner. Comparing to the existed protocols, our indole syntheses can be achieved at lower reaction temperature, in shorter reaction time, and with improved substrate tolerance.
Effect of commitments to catalysis on the degree of curvature in proton inventories of the kinetic parameters for enzyme-catalyzed reactions: Application to tryptophan indole-lyase
Kiick, Dennis M.
, p. 8499 - 8504 (1991)
A relatively simple method for obtaining Dk, the intrinsic solvent deuterium isotope effect for an enzyme-catalyzed reaction, is presented and discussed. Steady-state and pre-steady-state determinations of the solvent deuterium and substrate deuterium isotope effects, the later obtained in both H2O and D2O, enable the development of a catalytic mechanism. The interrelatedness of the isotope effect equations for the enzyme system allows calculation of Dk. As a result, the variation of l/Dka, the reciprocal of the partial intrinsic solvent deuterium isotope effect, with n(D2O), the atom fraction of deuterium in mixed isotopic waters, has been determined for the tryptophan indole-lyase enzyme-catalyzed reaction, and is compared to the variation of (V/K)n/(V/K)0 with n(D2O). Comparison of the proton inventories indicates that without knowledge of the commitments to catalysis underestimates of the number of protons calculated from a fit of the kinetic parameter data to the Gross-Butler equation can occur. A series of curves is presented which demonstrate that the degree of curvature in a bowl-shaped proton inventory decreases as the commitment to catalysis increases, and that ultimately, as the relative size of the commitment continues to increase, a dome-shaped proton inventory will result.
Efficient nickel-mediated intramolecular amination of aryl chlorides
Omar-Amrani, Rafik,Thomas, Antoine,Brenner, Eric,Schneider, Raphael,Fort, Yves
, p. 2311 - 2314 (2003)
(Matrix presented) The use of an in situ generated Ni(0) catalyst associated with 2,2′-bipyridine or N,N′ -bis(2,6-diisopropylphenyl)dihydroimidazol-2-ylidene (SIPr) as a ligand and NaO-t-Bu as the base for the intramolecular coupling of aryl chlorides with amines is described. The procedure has been applied to the formation of five-, six-, and seven-membered rings.
Visible light-mediated decarboxylative amination of indoline-2-carboxylic acids catalyzed by Rose Bengal
Zhang, Meng-Jie,Schroeder, Griffin M.,He, Yan-Hong,Guan, Zhi
, p. 96693 - 96699 (2016)
A visible light-induced decarboxylative amination of indoline-2-carboxylic acids and azodicarboxylate esters has been developed, in which the oxidation of α-amino acids provides a versatile CO2-extrusion platform to generate α-aminoalkyl radicals. The corresponding products were obtained with yields of up to 72% catalyzed by a metal-free photocatalyst under mild conditions.
A preparative synthesis of indole by dehydrogenation of 4,5,6,7-tetrahydroindole over catalysts with a low palladium content
Ryashentseva, M. A.
, p. 1756 - 1757 (1993)
Catalysts containing 0.15 - 0.5 percent of Pd are highly active and selective in the dehydrogenation of 4,5,6,7-tetrahydroindole to indole when γ-Al2O3 or Sibunite are used as supports. - Keywords: Pd-containing catalysts, tetrahydroindole, indole, dehydrogenation.
Turning tryptophanase into odor-generating biosensors
Xu, Yaqin,Zhang, Zhuyuan,Ali, M. Monsur,Sauder, Joanna,Deng, Xudong,Giang, Karen,Aguirre, Sergio D.,Pelton, Robert,Li, Yingfu,Filipe, Carlos D. M.
, p. 2620 - 2622 (2014)
An odor-based sensor system that exploits the metabolic enzyme tryptophanase (TPase) as the key component is reported. This enzyme is able to convert an odorless substrate like S-methyl-L-cysteine or L-tryptophan into the odorous products methyl mercaptan or indole. To make a biosensor, TPase was biotinylated so that it could be coupled with a molecular recognition element, such as an antibody, to develop an ELISA-like assay. This method was used for the detection of an antibody present in nM concentrations by the human nose. TPase can also be combined with the enzyme pyridoxal kinase (PKase) for use in a coupled assay to detect adenosine 5-triphosphate (ATP). When ATP is present in the low μM concentration range, the coupled enzymatic system generates an odor that is easily detectable by the human nose. Biotinylated TPase can be combined with various biotin-labeled molecular recognition elements, thereby enabling a broad range of applications for this odor-based reporting system. The nose knows: Tryptophanase (TPase), which converts S-methyl-L-cysteine into methyl mercaptan (smelly), was coupled to a molecular recognition element (such as an antibody) to create an odor-based biosensor. Biotinylated TPase could be combined with various biotin-labeled molecular recognition elements, thereby enabling a broad range of applications for this odor-based reporting system.
Ordered Porous Nitrogen-Doped Carbon Matrix with Atomically Dispersed Cobalt Sites as an Efficient Catalyst for Dehydrogenation and Transfer Hydrogenation of N-Heterocycles
Han, Yunhu,Wang, Ziyun,Xu, Ruirui,Zhang, Wei,Chen, Wenxing,Zheng, Lirong,Zhang, Jian,Luo, Jun,Wu, Konglin,Zhu, Youqi,Chen, Chen,Peng, Qing,Liu, Qiang,Hu,Wang, Dingsheng,Li, Yadong
, p. 11262 - 11266 (2018)
Single-atom catalysts (SACs) have been explored widely as potential substitutes for homogeneous catalysts. Isolated cobalt single-atom sites were stabilized on an ordered porous nitrogen-doped carbon matrix (ISAS-Co/OPNC). ISAS-Co/OPNC is a highly efficient catalyst for acceptorless dehydrogenation of N-heterocycles to release H2. ISAS-Co/OPNC also exhibits excellent catalytic activity for the reverse transfer hydrogenation (or hydrogenation) of N-heterocycles to store H2, using formic acid or external hydrogen as a hydrogen source. The catalytic performance of ISAS-Co/OPNC in both reactions surpasses previously reported homogeneous and heterogeneous precious-metal catalysts. The reaction mechanisms are systematically investigated using first-principles calculations and it is suggested that the Eley–Rideal mechanism is dominant.
An Equilibrium and Calorimetric Investigation of the Hydrolysis of L-Tryptophan to (Indole + Pyruvate + Ammonia)
Tewari, Yadu B.,Goldberg, Robert N.
, p. 167 - 184 (1994)
Apparent equilibrium constants and calorimetric enthalpies of reaction have been measured for the reaction L-tryptophan(aq) + H2O(l) = indole(aq) + pyruvate(aq) + ammonia(aq) which is cataluzed by L-tryptophanase.High-pressure liquid-chromatography and microcalorimetery were used to perform these measurements.The equilibrium measurements were performed as a function of pH, temperature, and ionic strength.The results have been interpreted with a chemical equilibrium model to obtain thermodynamic quantities for the reference reaction: L-tryptophan(aq) + H2O(l)= indole(aq) + pyruvate-(aq) + NH4+(aq).At T=25 deg C and Im=0 the results for this reaction are: K0=(1.05+/-0.13)x10E-4, ΔrG0=(22.71+/-0.33) kJ-mol-1, ΔrH0=(62.0+/-2.3) kJ-mol-1, and ΔrS0=(132+/-8) J-K-1-mol-1.These results have been used together with thermodynamic results from the literature to calculate standard Gibbs energies of formation, standard enthalpies of formation, standard molar entropies, standard molar heat capacities, and standard transformed formation properties for the substances participating in this reaction.
Electrochemical Dearomative Dicarboxylation of Heterocycles with Highly Negative Reduction Potentials
Hayashi, Hiroki,Kanna, Wataru,Maeda, Satoshi,Mita, Tsuyoshi,Takano, Hideaki,You, Yong
supporting information, p. 3685 - 3695 (2022/03/08)
The dearomative dicarboxylation of stable heteroaromatics using CO2is highly challenging but represents a very powerful method for producing synthetically useful dicarboxylic acids, which can potentially be employed as intermediates of biologically active molecules such as natural products and drug leads. However, these types of transformations are still underdeveloped, and concise methodologies with high efficiency (e.g., high yield and high selectivity for dicarboxylations) have not been reported. We herein describe a new electrochemical protocol using the CO2radical anion (E1/2of CO2= -2.2 V in DMF and -2.3 V in CH3CN vs SCE) that produces unprecedented trans-oriented 2,3-dicarboxylic acids from N-Ac-, Boc-, and Ph-protected indoles that exhibit highly negative reduction potentials (-2.50 to -2.94 V). On the basis of the calculated reduction potentials, N-protected indoles with reduction potentials up to -3 V smoothly undergo the desired dicarboxylation. Other heteroaromatics, including benzofuran, benzothiophene, electron-deficient furans, thiophenes, 1,3-diphenylisobenzofuran, and N-Boc-pyrazole, also exhibit reduction potentials more positive than -3 V and served as effective substrates for such dicarboxylations. The dicarboxylated products thus obtained can be derivatized into useful synthetic intermediates for biologically active compounds in few steps. We also show how the dearomative monocarboxylation can be achieved selectively by choice of the electrolyte, solvent, and protic additive; this strategy was then applied to the synthesis of an octahydroindole-2-carboxylic acid (Oic) derivative, which is a useful proline analogue.
A biomass-derived N-doped porous carbon catalyst for the aerobic dehydrogenation of nitrogen heterocycles
Cui, Fu-Jun,Guo, Fu-Hu,Liu, Jing-Jiang,Liu, Xiao-Yu,Quan, Zheng-Jun,Ullah, Arif,Wang, Xi-Cun,Zhu, Ji-Hua
supporting information, p. 1791 - 1799 (2022/01/31)
N-doped porous carbon (NC) was synthesized from sugar cane bagasse, which is a sustainable and widely available biomass waste. The preferred NC sample had a well-developed porous structure, a graphene-like surface morphology and different N species. More
Chemoselective hydrosilylation of carboxylic acids using a phosphine-free ruthenium complex and phenylsilane
Abhilash, Vishwanathan,Gadakh, Amol V.,Ganesh, Sambasivam,Hegde, Shivaprasad N.,Jacob, Anand,Karthik, C. S.,Lamees, Thundianandi,Mathivanan, Namachivayam,Sathiyanarayanan, Arumugam Murugan
supporting information, (2022/03/01)
A highly chemoselective hydrosilylation of carboxylic acids was achieved using a bench-stable, phosphine-free Ru-complex tethered with hemi-labile thiophene ligands as the catalyst, employing phenylsilane as the reducing agent. The methodology was further elaborated towards the one-pot synthesis of indole and benzoxazine via tandem reduction/cyclization of acid and nitro group.
Nickel-Catalyzed Addition of C–C Bonds of Amides to Strained Alkenes: The 1,2-Carboaminocarbonylation Reaction
Ito, Yuri,Kodama, Takuya,Nakatani, Syun,Shiraki, Ryota,Tobisu, Mamoru
supporting information, p. 662 - 666 (2022/02/05)
C(aryl)–C(═O) bonds of aryl amides can be activated and added across alkenes with the aid of a nickel catalyst. This 1,2-carboaminocarbonylation reaction enables the dicarbofunctionalization of alkenes with an atom economy of 100%.